Publié le
Lecture 10 mins
Postconditionnement : protéger le myocarde ischémique à la reperfusion
M. COUR, L. ARGAUD, Inserm U886 Cardioprotection, service de réanimation médicale ; groupement hospitalier Edouard-Herriot, Lyon, J. LOUFOUAT, M. OVIZE, Inserm U886 Cardioprotection, Lyon
L'infarctus du myocarde (IDM) demeure la première cause de décès dans le monde, alors même que le développement des unités de soins intensifs et des procédures de reperfusion a considérablement réduit sa mortalité précoce. Les conséquences de l'IDM en termes de morbi-mortalité et de pronostic fonctionnel sont directement liées à la taille de l'infarctus, qui est certes due à la durée de l'occlusion coronaire, mais aussi paradoxalement à l'importance des lésions myocardiques consécutives à la reperfusion. Des interventions thérapeutiques adjuvantes de la reperfusion coronaire, connues sous le nom de postconditionnement, sont aujourd'hui disponibles au laboratoire pour protéger le myocarde des conséquences de la reperfusion. Les premières applications cliniques de ces travaux offrent de réelles perspectives d'amélioration de la prise en charge des patients à la phase aiguë de l'IDM. Et au-delà, l'ensemble des pathologies cardiovasculaires mettant en jeu un mécanisme physiopathologique d'ischémie-reperfusion pourrait bénéficier à terme de cette recherche translationnelle.
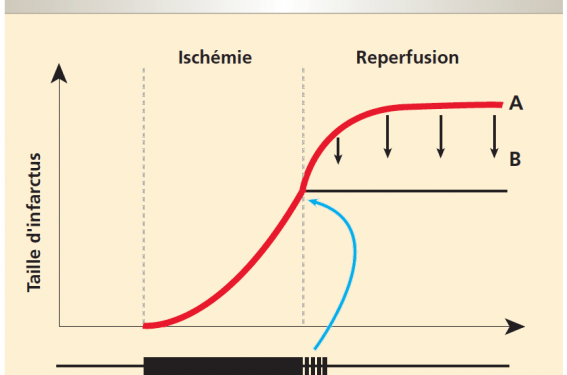
Nécrose de reperfusion Jusqu’à récemment, la reperfusion n’était envisagée que comme un phénomène bénéfique capable de « sauver » le myocarde ischémique et ainsi stabiliser la taille de l’infarctus à un niveau déterminé par la fin de l’ischémie. Nous savons aujourd’hui que la reperfusion est, par...
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



